 Arelis Agosto
Arelis AgostoDrug development pipelines are the lifeblood of the pharmaceutical industry. It is crucial for investors to understand milestones like FDA approvals and the impact of losing exclusivity on profits to navigate the dynamic health care sector. This piece provides an investment-focused overview of a typical drug’s lifecycle.
This piece is Part 1 in a 2-part series on the fundamentals of investing in pharmaceutical companies. The second part will delve into the key characteristics that can help inform an investigational treatment’s success.
Key Takeaways
- The regulatory process for investigational drugs is long and costly, but the rewards for successfully bringing a new drug to market can be vast.
- There are numerous potential pathways to accelerate the drug discovery process, which may notably impact a new drug’s return on investment.
- Generic competition can quickly eat into sales and reduce profit margins, leading investors to keep a close eye on new drug development.
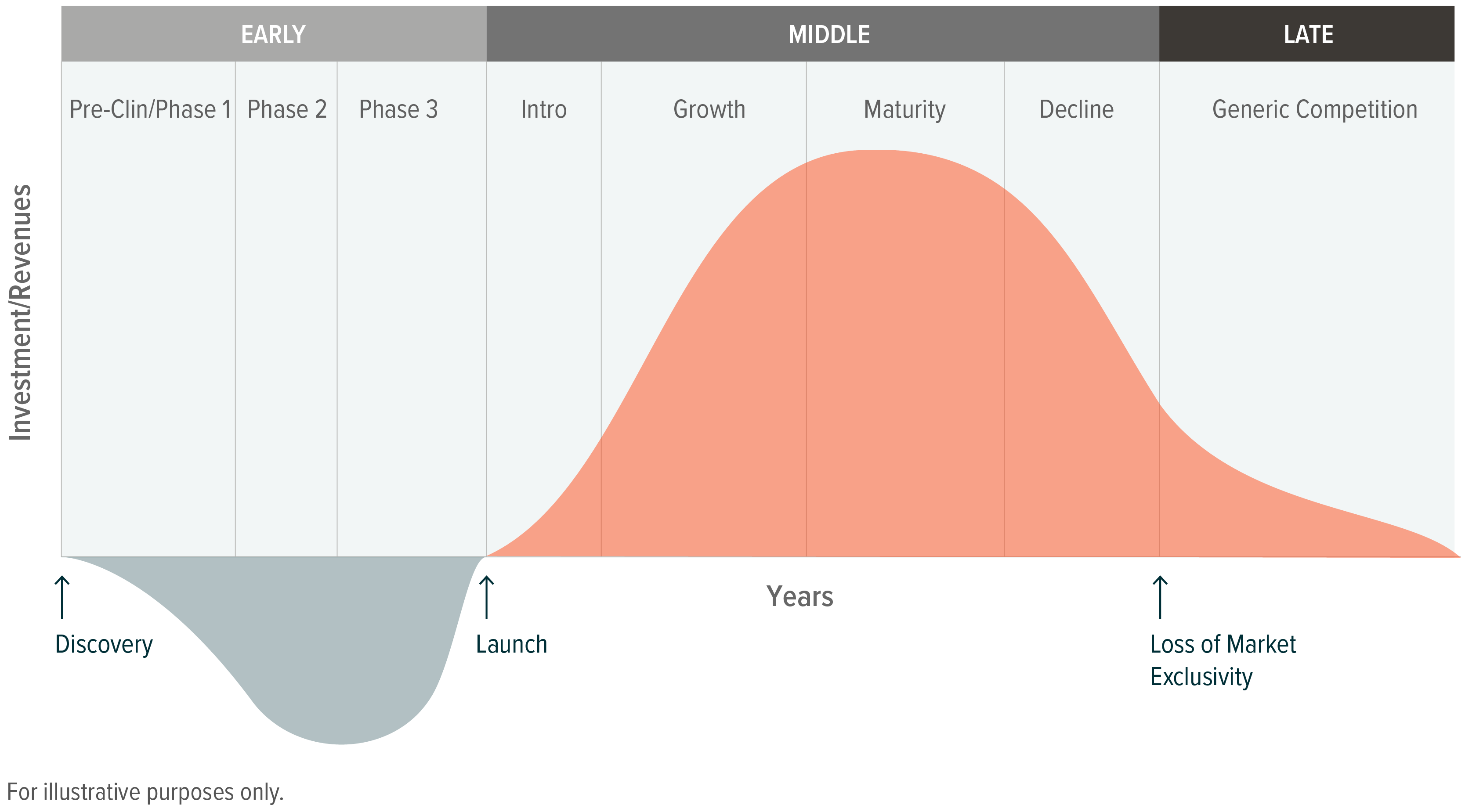
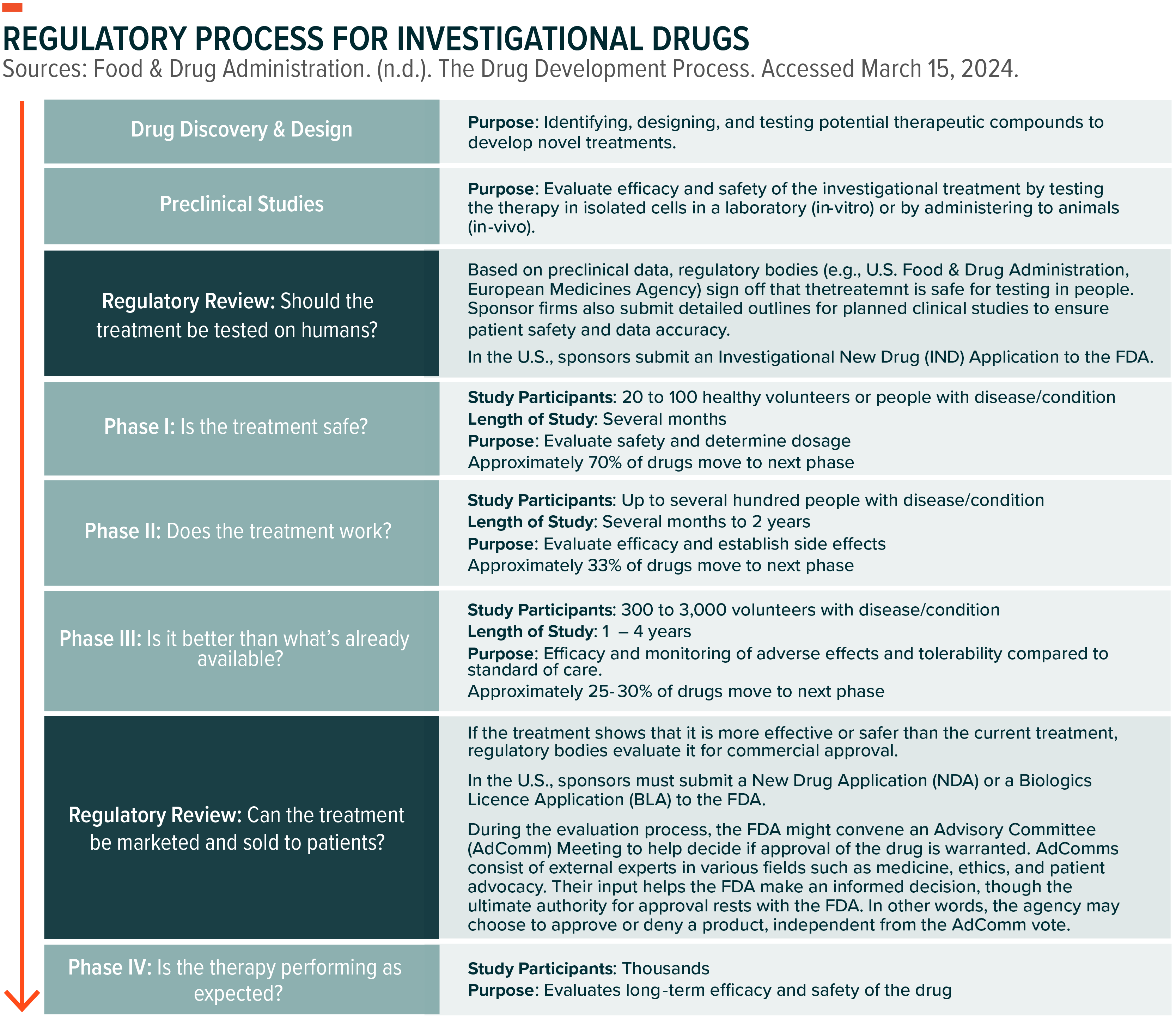
Drug Discovery & Development Process of an Investigational Drug
Investigational new drugs go through a rigorous process to ensure efficacy and safety. In the U.S., this process takes, on average, 10 to 15 years to ensure approved treatments are beneficial and safe for patients.1 Key regulatory steps include:
Pathways to Accelerate the Drug Development Process
Though the process for all investigational drugs is largely the same, regulatory bodies have outlined potential accelerated pathways to speed up the availability of drugs that treat serious diseases.2 When granted, these decisions can notably impact the return on investment for investigational drugs and can thus have a significant impact on biotech stock prices.3 The U.S. FDA has four key such designations:4
- Fast Track: Designed to streamline the development and review process of drugs targeting serious conditions with high unmet medical needs. Among other things, pharmaceutical sponsors have more frequent interactions with the FDA throughout the development process and can submit regulatory filings on a rolling basis.
- Breakthrough Therapy: Designed to streamline the development and review process of drugs that may demonstrate substantial improvement over available therapies. Like Fast Track designations, this process allows for increased interactions with the FDA and rolling reviews.
- Accelerated Approval: Designed to accelerate the development process of drugs that address illnesses with high unmet medical needs. This pathway allows for greater flexibility in evidence requirements for approval, as many illnesses (e.g., Alzheimer’s) progress slowly and waiting for clinical outcomes data can take years.
- Allows clinical trials to utilize “surrogate endpoints” for clinical trials. Surrogate endpoints are markers or measurements that are considered reasonably likely to predict a real clinical benefit. These are common in cancer clinical trials, for example, where measuring tumor shrinkage can occur far sooner than waiting to learn if a patient lived longer.
- May also require fewer clinical trials upfront. These treatments can, for example, be conditionally approved after a phase II clinical trial. In these instances, a phase III clinical trial is typically required to confirm the drug’s effectiveness and safety. The FDA can then decide to grant full approval to the drug or, in instances where the confirmatory trial fails, to modify or withdraw the drug’s approval.
- Priority Review: Designed to accelerate the review process of investigational drugs that may demonstrate substantial improvement over available therapies. Once granted, the FDA agrees to take action on an application within six months, compared to 10 months under standard review.
Pharmaceutical sponsors can also, subject to FDA approval, design combined or hybrid clinical trials (e.g., phase 1/2, or phase 2/3) that can reduce the time and resources required for drug development. Combined trials can also allow for faster patient recruitment and more efficient utilization of data while collecting all the necessary data required for regulatory review. Regulatory agencies like the FDA closely review the clinical trial design and results to ensure they meet the necessary standards for approval.
Post-Approval Exclusivity: The Period of Peak Profitability
Once a treatment is approved, the drug receives market exclusivity for a certain period. Different types of products receive different exclusivity timelines, though in most cases, a five-year exclusivity is provided. The FDA can provide certain extensions to exclusivity, including treatments for orphan diseases (those with less than 200,000 patients in the U.S.) and treatments for pediatric illnesses. A treatment can also receive additional exclusivity after its approval if it receives subsequent approval for a new illness or in a new delivery form (e.g., a pill rather than an injection).
Loss of Exclusivity: Entry of Generic and Biosimilar Competition
Once the exclusivity period concludes, competition is allowed to enter the market, usually at a discounted price. These are equivalent products approved through abbreviated pathways. Depending on the technology, the equivalent product is referred to as a:
- Generic: For drugs with specific chemical structures, generics are created to be the same as an already approved brand-name product. The generic must be the same dosage form, strength, quality, and intended use to ensure the generic treatment works in the same way and provides the same benefit as the branded drug.
- Biosimilars: Biosimilars are highly similar to large molecule drugs – also referred to as biologics. Biologics are molecules derived from living organisms and include, for example, vaccines. Corresponding biosimilars have no clinically meaningful difference from the biologic, meaning they have the same effectiveness and safety profile. There are currently 46 approved biosimilars in the U.S.5
When equivalent products enter the market, prices for the drugs decrease significantly. A single generic competitor can lead to price reductions of 30%, while five competing generics can lead to price reductions of nearly 85%.6
Manufacturers of the branded drug face what is colloquially referred to as a “patent cliff” once a drug faces generic competition, as sales of the branded product may decline up to 90%.7 To replenish sales, pharmaceutical firms often rely on expected drug launches and mergers and acquisitions (M&A).

Conclusion
The pharmaceutical space is highly specialized, often relying on complex chemistry and biology. However, there are also some basic tenants of the drug discovery process that can be useful to investors when examining the industry, deciphering clinical updates, or understanding the catalysts behind stock-price movements.
Related ETFs
GNOM – Global X Genomics & Biotechnology ETF
AGNG – Global X Aging Population ETF
Click the fund name above to view current performance and holdings. Holdings are subject to change. Current and future holdings are subject to risk.